実空間密度汎関数法に基づいたCar-Parrinello分子動力学法の開発

小泉健一 こいずみ けんいち
東京大学大学院工学系研究科
Car-Parrinello (CP)分子動力学は1985年、物理分野を母体として誕生し、以来、その有効性を化学、生物分野まで広げながら発展してきた。この方法は量子論に基づいているため、化学結合の組み替えを必要とするシミュレーションを可能にする。しかし、計算時間の問題から現実的なモデルを用いた計算は難しい。固体表面の状態を精度良く再現するには1000~1万原子のシミュレーションを行う必要があり、これには超並列コンピュータの利用が必須になる。このような環境によく対応した実空間密度汎関数法(RSDFT)コードをベースにしたCP法を開発することにより、計算効率の飛躍的な向上をめざしている。
その第一段階として、安定した時間発展が可能なプログラムを作成することができた。現在、216原子サイズのシリコンを用いた50万ステップに及ぶ計算が安定して作動することを確認している。さらに、機能を付加し実際の物理量を算出することが可能となった。例えば、シリコンの拡散係数の値については、高温状態で平面波基底の結果をうまく再現できた。低温状態の精度についても確認中である。
東京大学物性研のスパコン上では、1000原子の測定で並列化効率が48%を超えている。しかしながら、高速化はまだ十分といえず、特に「京」での最適化が遅れている。分子動力学に用いられているルーチンの多くは並列化が十分ではなく、ここは未開拓の部分である。これから高速化を行っていきたい。
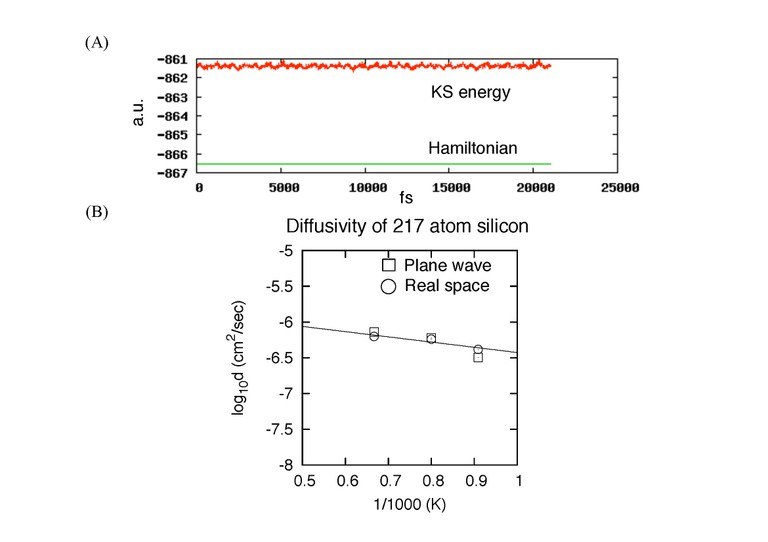 |
|
(A). 加熱10ps (1万step)、平衡化20ps、 測定20psのシリコン217原子(1原子は欠陥)におけるシミュレーションのエネルギー汎関数(KS-energy)とハミルトニアンの振る舞い。温度は1250K。保存量であるハミルトニアンは一定。 (B). 欠陥を含むシリコン結晶の拡散係数。1100K、1250K、1500Kで測定。実空間法の結果は、平面波基底での計算結果を良く再現す |