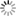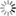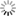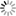Prediction of electron functions in nanostructures using the density functional approach
Interviewee:
|
Interviewer:
|
| Professor, Graduate School of Engineering, The University of Tokyo |
CMSI Condensed Matter Physics Division Researcher |
Overview of topic and objectives
The objective of this Priority Research Topic is to establish a high-speed calculation technique that makes it possible to use state-of-the-art supercomputers to perform first-principles calculations (using density functional theory) for nanostructures ranging in size from several tens of thousands to several hundreds of thousands of atoms. This will make it possible to determine the atomic and electron structure and device properties of these nanostructures and the mechanism by which the nanostructures are formed. This group is working closely with CMSI Research Group 1, which is studying more basic questions. Within CMSI Research Group 2, the role of this Priority Research Topic is the application of basic scientific knowledge to real-world technology. Achievements in the research for this
topic will make it possible to use first-principles calculations in quantum simulations, device simulations and other types of technology computer- aided design (TCAD).
In terms of actual calculations, two main methods are used for research and development: the Real-Space Density Functional Theory (RSDFT) and CONQUEST, an order-N first-principles application program. RSDFT introduces a grid into real space and calculates electron orbitals, electron density and potential and other quantities on grid points, and solves the Kohn-Sham equation. Unlike the density functional method that uses plane wave basis sets, this method does not require fast Fourier transformation (FFT), which uses communication between all CPUs. This method also makes it possible to set arbitrary boundary conditions for the wave function, such as nonperiodic system, periodic system and so on.
Rather than determining the eigenfunctions of the Kohn-Sham equation, CONQUEST determines a one-body density matrix. With the conventional algorithms, the computational complexity required for N atoms is proportional to N³, while it is merely proportional to N by the order-N method. The order-N method thus enables first-principles calculations for ultra-large scale systems that are made up of tens or hundreds of thousands of atoms or more.
Current state
RSDFT was optimized for a massively parallel multicore architecture and achieved an effective performance of 3.08 petaflops (execution efficiency 43.6%) in calculations for a 100,000 atom silicon nanowire conducted using 70% of the overall resources on the K computer. This achievement was awarded the ACM Gordon Bell Prize (Sustained Performance Prize) at SC11, the international conference on high-performance computing. However, it would be difficult to perform simulations for several hundred thousand atoms without occupying 70% of the total resources of the K computer, and so at present RSDFT is only suitable for calculating several tens of thousands of atoms. RSDFT's ability to perform highly efficient parallel calculations also enables it to handle with comparative ease systems consisting of several thousand atoms, something that would require an enormous
amount of time to calculate using other methods. RSDFT has successfully determined the charge injection energy in silicon nanodots consisting of several tens to several hundreds of thousands of atoms, the electron state of silicon nanowires, the interaction between carbon nanotubes and silicon interfaces, the structure of silicene (silicon analogue of graphene on thin film) and so on. We have also improved functionals and remedied the bandgap problem by introducing the hybrid exchange correlation energy.
Future anticipated benefits
We are approaching the limit of miniaturization of semiconductor devices, and research is being pursued into nanoscale devices that use the quantum effect. Accelerating first-principles calculation methods to the greatest degree possible in order to enable simulations of the quantum effect that controls nano-level phenomena will result in new device designs and guidelines. Specific benefits that are anticipated include predicting the structural stability and electronic functions of nanodots and nanowires (which are expected to become the initiating agents for next-generation devices), establishment of quantum theory and identification of device characteristics for the transport of electrons, heat and atoms for next-generation devices and nano-junctions, establishment of device simulator as basic technologies for the post-scaling era, and so on.
Impact and contribution tosociety
To stimulate the semiconductor industry in Japan, it will be essential to use first-principles calculations in semiconductor simulations, and to expand this effort to include prediction of device material functions, development of new functional materials, and the search for new nanostructures. Achieving increased speed for RSDFT and CONQUEST on supercomputers will also lead to the development of software designed for use on supercomputers. However, it will not be possible to achieve high-performance calculations by hardware evolution alone; software that can make effective use of this evolution will also be needed. For this reason, software will have to evolve along with supercomputers. This Priority Research Topic will make a contribution to Japan's progress and tradition of supercomputer technology.
 |
| Cross-section of a 10 nm diameter silicon (Si) nanowire, revealing modeling of the atomic-scale irregularities in the lateral surface produced during the wire formation process. In all, electron state calculations were conducted for 14,336 atoms. |
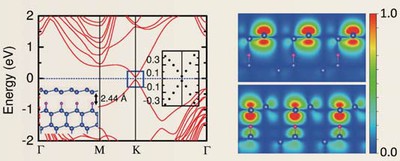 |
| Band structure of a silicon monolayer (silicene) with a honeycomb structure and density distribution of orbits near the Fermi level. The RSDFT calculations indicated that hydrotreating of the Si (111) surface resulted in the formation of a stable silicene film, and Dirac cones were produced near the Fermi level. |