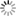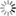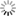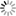Development of the molecular science of viruses through all-atom simulations
Interviewee:
|
Interviewer:
|
| Professor, Graduate School of Engineering, Nagoya University |
CMSI Molecular Science Division Researcher |
What is the reason for using the K computer to conduct virus simulations?
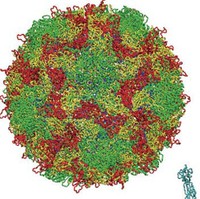
Protein simulations have often been conducted in the field of molecular science. Up to now, however, they've mostly been single proteins or proteins with ligands added. At most, they've been simulations of protein complexes consisting of several proteins. We wanted to try simulating enormous systems, in which many more proteins come together and, for the first time, have functions as life activities. From among the components of these enormous systems, we chose to study virus capsids. The virus capsid is the basic structural unit of a virus. In an all-atom model that includes water molecules, it consists of some ten million atoms. In order to simulate a ten million atom system, a supercomputer such as the K computer that has high computing
capacity is indispensable.
What special measures did you have to devise in order to use the K computer for this purpose?
In protein simulations, it's particularly important to accurately determine the long-range force. The Ewald method is one of the main methods used for accurate calculation of long-range force, but this method requires fast Fourier transformation (FFT). So the number of parallel nodes that can be used is limited to 200 to around 500, and this limits the applicable range of the system to several hundred thousand atoms.
To get around this problem, we did two things. The first was to introduce an algorithm called the fast multipole method (FMM) that is suited to parallel computing. It enables efficient parallelization even on a computer of the same class as the K computer with more than 80,000 nodes.
The other thing we did was to tune the program to match the K computer hardware. In particular, we simplified the data for large-scale parallelization, optimized communication for the 3-dimensional torus, took steps to reduce data transfer to and from memory (cache optimization), enhanced SIMD excecution and so on. This type of tuning was only possible through close communication with experienced computer science personnel at RIKEN and Fujitsu. As a result, even though our program (Modylas) is a general purpose utility, we achieved 41.1% of the theoretical peak performance and 5 milliseconds in actual time for each MD calculation step. We're currently continuing with the tuning process and working to provide the enhanced features needed to estimate free energy and other types of thermodynamic quantities.
What type of ripple effect do you anticipate that the achievements of research into this Priority Research Topic will have in the fields of molecular simulation and virology, and in society at large?
Viewed from the field of virology, our research is expected to lead to the discovery of antivirals and other drugs. Through simulations, we analyze the affinity between the virus and the receptors inside the cell membrane, and by showing the principle through which this is manifested, we will be able to propose new molecules that function as inhibitors and so on. We're also considering various other research topics, such as the affinity between viruses and antibodies, artificial vaccines and so on. We think that these research topics are important from the standpoint of society at large, since they relate directly to people's health.
In the field of molecular simulations, it will eventually become commonplace to perform large-scale simulations for systems with more than a million atoms. I think there will also be further progress in the methods used to handle the enormous quantities of data that will be obtained as a result, as well as analysis methods for thermodynamic quantities and so on. Large-scale systems are an area in which there are many possible research topics, not only in life science but in the fields of chemistry and materials as well. One example is the study of polymers, an area in which new material development is anticipated. In the future, you'll see studies of various polymers that could not be targeted up to now in the field of computational chemistry.
Finally, could you talk about the future prospects for this research?
One is the dissemination of our program. We're currently making preparations to provide the program to people who want to conduct simulations of large-scale systems. Another is a more careful study of the molecular theory of large-scale systems. We plan to conduct simulations of not only viruses but also other large-scale systems in the fields of life science, chemistry, materials and so on.
Our goal is to be able to more rigorously explain the principles that control the materials involved in these phenomena.
|
|
| The poliovirus and the D1 and D2 domains of the CD155 receptor. The water molecules used as a solvent have been removed from the image. (1 atmospheric pressure, 37°C) |