Development of Car-Parrinello molecular dynamics based on real-space density functional theory
Kenichi Koizumi
Graduate School of Engineering,
The University of Tokyo
Car-Parrinello molecular-dynamics (CPMD) method was developed by R. Car and M. Parrinello in 1985 and its effectiveness is now covering almost all fields of science such as physics, chemistry, and biology. CPMD is based on quantum theory and it can simulate the breaking and reformation of chemical bonds, which are impossible to be described by classical molecular dynamics. However, its effectiveness is still underestimated being hindered by huge computational costs. To simulate chemical reactions in device-scale system ncluding more than thousands of atoms, usage of the massive parallel computation is inevitable. Therefore, we are developing CPMD based on real-space density functional theory (RS-CPMD) that is a suitable method to extract the powers of the massively parallel computations. Now, we have developed a stable code enabling over 500,000 step simulations using 217 Si atoms and those results nicely reproduce those of a plane-wave code. However, there remain some rooms for improving the efficience because the optimization to the K computer is still insufficient. Now, we are trying to tune the code and will realize device-scale CPMD simulations that are difficult to perform by the standard plane-wave codes.
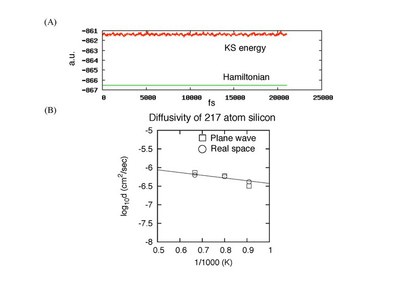 |
| (A) Kohn-Sham energy and Hamiltonian of the total 50ps simulation (500,000 steps) at 1250K. Heating is 10ps, equilibration is 20ps, and production run is 20ps. Note that our Hamiltonian is totally constant. (B) Diffusivities of crystalline silicon including an interstitial defect. The temperatures are 1100K, 1250K, and 1500K. The results of RS-CPMD reproduce those of a plane-wave code at high temperature. |